VQEProblem#
- class VQEProblem(hamiltonian, ansatz_function, num_params, init_function=None, callback=False)[source]#
Central structure to facilitate treatment of VQE problems. This class encapsulates the Hamiltonian, the ansatz, and the initial state preparation function for a specific VQE problem instance.
- Parameters:
- hamiltonianQubitOperator or FermionicOperator
The problem Hamiltonian.
- ansatz_functioncallable
A function receiving a QuantumVariable, and an array of real parameters of size
(n,). This function implements the unitary corresponding to one layer of the ansatz.- num_paramsint
The number of parameters \(n\) per layer of the ansatz.
- init_functioncallable, optional
A function receiving a QuantumVariable and preparing the initial state from the \(\ket{0}\) state. By default, the inital state is the \(\ket{0}\) state.
- callbackbool, optional
If
True, intermediate results are stored. The default isFalse.
Examples
For a quick demonstration, we show how to calculate the ground state energy of the \(H_2\) molecule using VQE. The
electronic_structure_problemmethod generates a VQEProblem instance with the Hamiltonian and a chemistry-inspired Qubit Coupled Cluster Single Double (QCCSD) ansatz.from pyscf import gto from qrisp import QuantumVariable from qrisp.vqe.problems.electronic_structure import * mol = gto.M( atom = '''H 0 0 0; H 0 0 0.74''', basis = 'sto-3g') vqe = electronic_structure_problem(mol) energy = vqe.run(QuantumVariable(4), depth=1, max_iter=50) print(energy) #Yields -1.8461290172512965
You can also specify a custom Hamiltonian and a custom hardware-efficient ansatz, as explained here.
from qrisp import * from qrisp.operators.qubit import X,Y,Z # Problem Hamiltonian c = [-0.81054, 0.16614, 0.16892, 0.17218, -0.22573, 0.12091, 0.166145, 0.04523] H = c[0] \ + c[1]*Z(0)*Z(2) \ + c[2]*Z(1)*Z(3) \ + c[3]*(Z(3) + Z(1)) \ + c[4]*(Z(2) + Z(0)) \ + c[5]*(Z(2)*Z(3) + Z(0)*Z(1)) \ + c[6]*(Z(0)*Z(3) + Z(1)*Z(2)) \ + c[7]*(Y(0)*Y(1)*Y(2)*Y(3) + X(0)*X(1)*Y(2)*Y(3) + Y(0)*Y(1)*X(2)*X(3) + X(0)*X(1)*X(2)*X(3)) # Ansatz def ansatz(qv,theta): for i in range(4): ry(theta[i],qv[i]) for i in range(3): cx(qv[i],qv[i+1]) cx(qv[3],qv[0]) from qrisp.vqe.vqe_problem import * vqe = VQEProblem(hamiltonian = H, ansatz_function = ansatz, num_params = 4, callback = True) energy = vqe.run(QuantumVariable(4), depth = 1, max_iter = 50) print(energy) # Yields -1.864179046
Note that for comparing to the results in the aforementioned paper, we have to add the nuclear repulsion energy \(E_{\text{nuc}}=0.72\) to the calculated electronic energy \(E_{\text{el}}\).
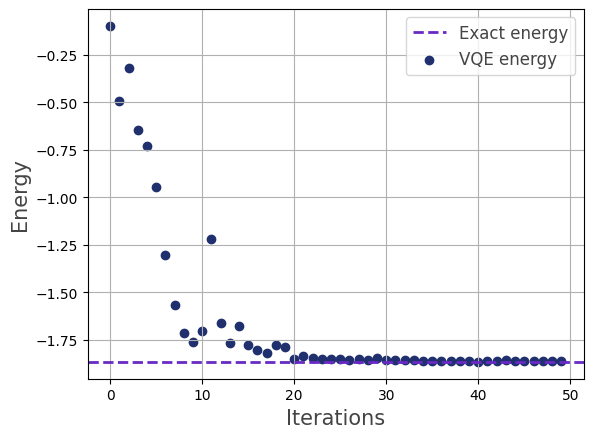
We visualize the optimization process:
>>> vqe.visualize_energy(exact=True)
Methods#
|
Set the initial state preparation function for the VQE problem. |
|
Run VQE for the specific problem instance. |
|
This function allows for training of a circuit with a given instance of a |
|
Compiles the circuit that is evaluated by the |
|
This method enables convenient data collection regarding performance of the implementation. |
|
Visualizes the energy during the optimization process. |